¿QUE ES LA BAJA VISIÓN?
Baja visión es la condición visual que padece una persona con una reducción importante de su visión, que no mejora utilizando la adecuada corrección en gafas, lentes de contacto e incluso acertados tratamientos farmacológicos o cirugía, y que por ello sufre una incapacidad para realizar algunas tareas de la vida cotidiana
Se considera que un paciente tiene baja visión cuando tras la mejor corrección óptica, su Agudeza visual es menor de 0,3 en el mejor de los ojos, o un campo visual inferior a 30º
Esta incapacidad ó limitación puede tener su origen en diferentes patologías, habitualmente relacionadas con la edad, como pueden ser, Degeneración Macular, Retinopatía Diabética, Retinosis Pigmentaria, Glaucoma, etc.
La finalidad de la baja visión es potenciar éste resto visual mediante el uso de ayudas especiales que permitirán recuperar cierta funcionalidad para las actividades abandonadas.
ALGUNAS PATOLOGIAS QUE PROVOCAN BAJA VISION
¿Que es la Retinopatía Diabética?
Las manifestaciones de la diabetes a nivel ocular son numerosas y complejas, pudiendo afectar a cualquier parte del sistema visual. La retina es la estructura afectada con mayor frecuencia e importancia, pero tanto el segmento anterior del ojo (córnea, cristalino, iris), como el nervio óptico y los nervios oculomotores pueden verse afectados.
Las cataratas son 1,6 veces más frecuentes en la población diabética, ocurren en edades más tempranas y progresan más rápidamente que en la población no diabética.
El glaucoma de ángulo abierto es 1,4 veces más frecuente en los diabéticos.
Después de 20 años del diagnóstico de diabetes, prácticamente el 100% de los pacientes con diabetes tipo 1 (DM1) y el 60% de los pacientes con diabetes tipo 2 (DM2) presentan retinopatía diabética. Ésta es la causa más frecuente de ceguera en los países industrializados. La población diabética tiene 25 veces más riesgo de ceguera que la población no diabética. Es la tercera patología causante de deficiencia visual entre los afiliados de la O.N.C.E.
La presencia de retinopatía, incluso en sus formas más agresivas con grave riesgo de ceguera, no suele provocar síntomas en la agudeza visual y cuando aparece una disminución en la misma, suele ser demasiado tarde para que pueda realizarse un tratamiento eficaz.
La detección precoz y el tratamiento adecuado, reduce más de un 60% el riesgo de pérdida visual en los pacientes con retinopatía diabética de alto riesgo y más de un 50% en caso de edema macular diabético.
Estudios recientes indican que un correcto control glucémico y de la tensión arterial pueden prevenir o retrasar la aparición de edema macular y retinopatía diabética proliferativa.
La compensación con ayudas ópticas especiales en los casos de Baja Visión, es la alternativa de elección en cuanto a lo que supone de recuperación funcional para el paciente con ésta grave afectación ocular.
 | |  |
¿Qué es DMAE?
La degeneración macular asociada a la edad es la causa más común de ceguera legal en personas de más de 50 años en los países occidentales.
La DMAE es una enfermedad ocular degenerativa que afecta a un área de la retina de su ojo llamada mácula. Ésta es la responsable de la visión central directa, necesaria para las actividades de la vida diaria como leer, ver la televisión o identificar a las personas.
Síntomas
- Visión borrosa en el campo central de visión, conservando la visión periférica.
- La percepción de las líneas rectas puede estar distorsionada y parecer líneas torcidas, inclinadas o desaparecer en algún punto.
- Punto negro en el centro del campo central.
- Deslumbramiento, falta de luz en la lectura.
(ver imágenes DMAE 2010)
Tipos
Existen dos tipos de degeneración de la mácula: húmeda y seca.
Forma seca
Tiene también una evolución más lenta que la húmeda. En sus inicios causará una visión ligeramente borrosa en la zona central y distorsionada. A medida que la enfermedad progresa, esta zona se va haciendo más grande y más borrosa.
Forma húmeda
La forma húmeda de esta afección conduce a una pérdida de visión más importante y más rápida que la seca. La forma húmeda de la DMAE es susceptible de ser tratada con láser o nuevos fármacos.
El tratamiento de la Degeneración Macular Asociada a la Edad está evolucionando y avanzando de forma rápida. Existen diferentes tratamientos para la DMAE, pero hay que señalar que, en ningún caso, van a curar la enfermedad, sino que pueden detenerla o retrasar sus avances.
En el caso de la DMAE húmeda y, especialmente en sus fases iniciales, los tratamientos clínicos que se aplican son el láser, la terapia fotodinámica y más recientemente, la aplicación intraocular de tres fármacos.
En ninguno de los casos estos medicamentos curan la enfermedad ni hacen que se recupere completamente la visión, pero se ha demostrado que cerca del 95% detienen la progresión de la enfermedad y el 40% de los enfermos tratados no sólo dejan de perder vista sino que incluso mejoran sus síntomas.
Pasos a seguir si tiene DMAE
1-Acudir a un oftalmólogo especialista en retina para que estabilice la enfermedad.
2-Acudir a un especialista en baja visión que le ayudara a potenciar su resto visual y le adaptará las ayudas ópticas necesarias para poder continuar realizando su vida normal.
Prevención
Es muy importante cuidar la visión, proteger los ojos, vigilar la evolución de la enfermedad en el caso de la forma húmeda para acudir al oftalmólogo ante un agravamiento de la enfermedad, y tomar vitaminas A, C, E y minerales.
1-Alimentación
Una alimentación rica en vegetales como zanahorias, repollo, espinacas, brócoli, guisantes, coles de Bruselas, maíz, etc. puede prevenir la pérdida de visión. O, por el contrario, una dieta pobre - por ejemplo, la denominada comida basura - puede favorecer la degeneración de la mácula.
La luteína y la zeaxantina, dos carotenoides que son pigmentos maculares, desempeñan un papel determinante en la protección de los ojos.
2- Gafas de sol filtrando la luz azul
Investigaciones científicas prueban que ambos componentes actúan como unas “gafas de sol internas” filtrando la luz azul y protegiéndolos de los radicales libres. Estos carotenoides de la retina son los principales componentes del sistema de defensa del ojo frente a la radiación solar.
3- Vitaminas
Investigaciones sobre la mácula, como el LUNA study¹, han mostrado la efectividad para la salud visual de la siguiente dosis diaria de micronutrientes
• Luteína 12 mg.
• Zeaxantina: 1 mg.
• Vitamina C: 120 mg
• Vitamina E: 17,6 mg
• Selenio: 40 µg
• Zinc. 10 mg.
Factores de riesgo
1- El primero es la edad. La DMAE puede aparecer en torno a los 50 años, aunque en un porcentaje muy reducido. A medida que la edad avanza, su incidencia se hace mucho mayor y puede decirse que la mitad de los europeos mayores de 65 años la padecen. Además de la edad hay otros factores de riesgo como:
2-La predisposición familiar. Las personas en cuya familia haya habido casos de DMAE, tienen más probabilidades de contraer la enfermedad.
3-Una dieta inadecuada pobre en luteína y zeaxantina. La carencia de vitaminas C y E, de zinc, así como de los dos carotenoides (luteína y zeaxantina) responsables del pigmento macular, afecta negativamente a su sistema de defensa y puede provocar la progresiva degeneración de la mácula.
4-El tabaco. El tabaco incrementa la producción de radicales libres que provocan un daño celular y pueden causar una escasa circulación hasta la retina de aquellos nutrientes que protegen a la mácula.
5-La obesidad. Investigaciones recientes muestran que el peso excesivo puede contribuir a una evolución más rápida de la enfermedad.
6-La exposición directa a la luz del sol, sin la protección de gafas apropiadas puede dañar la retina y motivar una pérdida de visión irreversible. La radiación de los rayos solares origina sustancias oxidantes dañinas, denominadas radicales libres, que afectan a la retina.
7-La hipertensión.
¿Que es el Glaucoma?
Más exactamente, la neuropatía optica glaucomatosa (NOG) es una enfermedad del nervio óptico caracterizada por la pérdida adquirida de fibras nerviosas, de carácter progresivo que desencadena la atrofia irreversible de las mismas. La consecuencia define una amaurosis ó ceguera gradual pero continua, que penaliza e incapacita de forma escalonada pero inexorable el normal desenvolvimiento diario en las tareas más cotidianas.
La NOG cuyo resultado final es la pérdida de campo visual y la atrofia de la cabeza del nervio óptico, continúa siendo aún una enfermedad de etiopatogenia incierta, si bien se conocen bastantes factores que la provocan. En el glaucoma primario de ángulo abierto (GPAA), la referencia más conocida y estudiada es la elevación de la presión intraocular (PIO), que sin menospreciar otros factores de riesgo, el objetivo principal del tratamiento va encaminado a reducir los valores de la misma.
Aunque la NOG tiene una alta prevalencia en el mundo, la variación de los datos obtenidos dependerá de la estimación referida al estudio que consultemos, si bien en todos se constata como la segunda causa de ceguera en todo el mundo.
Por tratarse de una enfermedad asintomática, casi dos tercios de los pacientes afectos no notarán una reducción ó pérdida de agudeza visual hasta que no se llega a una fase avanzada de la misma, razón por la cual gran parte de éstos enfermos son diagnosticados tardíamente.
El resultado se refleja en una grave alteración funcional asociada a la lesión de axones y posterior muerte de células ganglionares en la retina, como consecuencia de éste cuadro, se produce una pérdida funcional del campo visual periférico y un deterioro general de la visión. Todo ello va lastrando de manera importante la movilidad y la capacidad de realizar tareas cotidianas como desplazarse de un lugar a otro o incluso leer, actividades para las que resulta imprescindible disponer de una buena visión dinámica. En éste sentido, el entrenamiento motor y la rehabilitación visual con ayudas especiales para baja visión resulta necesario para restablecer el control funcional de determinadas tareas.
SUBIR
¿Qué es la Miopía Magna?
- Es la miopía producida por una elongación excesiva del globo ocular (eje antero posterior mayor de 26 mm.).
- Se inicia en la infancia y suele progresar en la vida adulta.
- Dependiendo de la severidad de la miopía magna pueden aparecer cambios degenerativos asociados a la elongación excesiva del ojo, especialmente a nivel de la retina y el polo posterior del ojo (retinopatía miópica).
- El error refractivo (número de dioptrías) es normalmente superior a 14 dioptrías.
- En ocasiones se producen alteraciones vítreo retinianas, desprendimiento de retina, inflamación o hemorragia macular, degeneración retiniana, atrofia coroidal, cataratas, y glaucoma que pueden originar complicaciones y comprometer seriamente la visión de la persona con miopía.
- Se considera una enfermedad ocular y por tanto debe controlarse periódicamente por el oftalmólogo o el optometrista, que además de graduar la vista tendrá que hacer una exploración ocular completa, incluyendo el fondo de ojo para examinar la retina, y detectar precozmente alguna lesión que pueda tratarse y de este modo evitar complicaciones futuras.
Síntomas
La condición de la miopía patológica afecta principalmente a las personas de los 30 a 40 años de edad y puede dar lugar a graves impedimentos visuales o incluso la extrema pérdida de la visión.
La distorsión de la visión central es uno de los primeros síntomas de la miopía patológica, líneas rectas puede parecer en realidad doblados o distorsionada, percepción frecuente de moscas volantes Pueden experimentar cambio en su percepción del color, y tener además problemas con la sensibilidad al contraste.
La miopía patológica también puede incluir problemas con la lectura, ver la televisión, también pueden notar una mancha en el centro de su campo visual que aparece borroso, o en color gris y otros aspectos de la vida cotidiana, tales como la conducción.
El diagnóstico de la miopía patológica puede ocurrir a menudo durante un examen ocular de rutina. Si se considera que existe este tipo de miopía, el optometrista debe vigilar al paciente y en caso de complicaciones, como el desprendimiento de retina, derivar urgentemente al oftalmólogo, ya puede ser una de las complicaciones asociadas a la miopía patológica, aunque algunas zonas de visión periférica por lo general se mantienen, en función de la zona desgarrada.
Alrededor de dos por ciento de la población se ve afectada por la miopía patológica, y hasta hace poco, se creía que sólo las gafas con muchas dioptrías podían utilizarse para solucionarla. En los últimos años, no obstante, se han logrado buenos resultados con la medicación, dando esperanza para la mejora de la visión de las personas que sufren de la miopía patológica.
Factores de riesgo
Tener miopía magna no consiste únicamente en tener muchas dioptrías. Las personas con miopía magna tienen una mayor susceptibilidad de presentar complicaciones oculares. Se ha asociado a un mayor riesgo de presentar cataratas, glaucoma o alteraciones del polo posterior del ojo (atrofia corioretiniana, degeneración retiniana, desprendimiento de retina, alteraciones del disco óptico o degeneración macular).
Este riesgo es mayor cuanto más alongado está el globo ocular. Aunque muchas de estas alteraciones tienen tratamientos efectivos, en ocasiones las consecuencias de estas complicaciones pueden comprometer de forma importante la visión de la persona con miopía magna.
Prevención
El abordaje y tratamiento de las complicaciones de este tipo de miopías, así como los nuevos retos en investigación, son algunos de los aspectos que ocupan el interés en miopía magna. "Las lesiones de la mácula son las de mayor relevancia y, tal vez, las de mayor actualidad porque existen tratamientos que han permitido que la visión de los miopes magnos no sea tan deficiente".
Posibles abordajes
Se calcula que un 20 por ciento de la población española es miope y de ellos, entre el 1 y el 2 por ciento tienen miopía magna. Las lesiones maculares, además de ser las de mayor relevancia patológica, son también las que mayor interés clínico despiertan porque disponen de tratamientos con antiangiogénicos y con terapia fotodinámica que han supuesto un avance, ya que "gracias a ellos los afectados conservan una visión menos deficitaria".
La lesión de la mácula de la miopía magna tiene una expresión clínica, aunque un mecanismo de origen diferente, muy similar a la de la degeneración macular asociada a la edad (DMAE). El tratamiento es también parecido, aunque en miopía son mejores que en la DMAE.
La utilización del láser es uno de los abordajes más antiguos, si bien se emplea solamente para neovascularizaciones que están alejadas de la mácula. Cuando la lesión se localiza exactamente en la mácula se opta por la ya clásica fotodinámica, que básicamente consiste en la inyección de verteporfino que se deposita sobre las membranas vasculares que son destruidas con láser.
La alternativa que se ensaya actualmente es la terapia antiangiogénica. "Se basa en la inyección intraocular de estos agentes, que casi siempre hay que repetir un mínimo de tres veces dependiendo de la evolución, y con ella se impide el desarrollo de las neovascularizaciones en la zona de la mácula".
En este caso, la lesión se encuentra entre la retina y la coroides, lo que significa que "un abordaje muy temprano que evite el daño irreversible de la mácula permitiría mantener una visión relativamente efectiva. En estas patologías la clave del éxito se encuentra en la detección precoz".
Pasos a seguir si tiene Miopía Magna
1-Acudir a un oftalmólogo o especialista en retina para que realicen un fondo de ojo completo.
2-Acudir a un especialista en baja visión que le ayudara a potenciar su resto visual y le adaptará las ayudas ópticas necesarias para poder continuar realizando su vida normal.
| |  |
¿Qué es la Retinosis Pigmentaria?Se denomina Retinosis Pigmentaria (RP) a un conjunto de degeneraciones progresivas de carácter hereditario que, afectan principalmente a la función de los fotorreceptores y al epitelio pigmentario de la retina.
Epidemiología
En España el número de afectados supera los 15.000 individuos, estimándose en 480.000 los portadores del gen defectuoso y por tanto posibles transmisores de esta enfermedad. La RP es la distrofia retiniana más frecuente. Afecta a un 1/5000 de la población mundial. y es la cuarta causa más frecuente de ceguera en el mundo. Es ligeramente más frecuente en varones, 55-63% lque en mujeres.
Síntomas
- Ceguera nocturna; mala adaptación a la oscuridad.
- Campo de visión limitado: pérdida de visión periférica. Para poder ver los objetos que están a su alrededor tienen que girar la cabeza (visión "en túnel").
- Deslumbramiento: necesario gafas de sol especiales.
- Es frecuente la miopía.
- También se han descrito casos asociados a una hipermetropía severa.
- Evoluciona de manera diferente según las personas y no conducen todas a la misma pérdida de visión.
Medidas preventivas
-Alimentación
- Evitar en lo posible alimentos industrializados: conservantes, aromatizantes, colorantes, que se encuentran en los alimentos enlatados y procesados.
- Cocinar con sal común y añadir ajo, cebolla y perejil, si gusta.
- Dieta nula en: alcohol, café y derivados y tabaco; y limitada en bebidas refrescantes.
- No consumir golosinas o empaquetados dulces o salados (patatas fritas, cacahuetes...).
- Dieta rica en ácidos grasos poliinsaturados: aceite de oliva, pescados azules como el salmón, sardinas, chicharro, anchoas, etc. 2-3 veces por semana.
- Dieta limitada en grasas saturadas (animales, quesos, mantequilla, etc.) o sospechosos de tener grasas animales (repostería, precocinados, etc.) o Poliinsaturadas hidrogenadas (margarinas). Azúcar (glucosa, sacarosa, lactosa, dextrosas, etc.) y cualquier alimento que lo contenga. Alimentos refinados, industrializados y procesados (fritos, etc.).
- Consumir hortalizas y ensaladas, especialmente el tomate y la zanahoria en zumos, triturado, rallado, cocinado.
- Consumir abundante fruta al día fundamentalmente del tiempo: naranja, uva negra, albaricoque, manzana, limón, etc.
- Consumir muy frecuentemente legumbres.
- El pan, arroz y cereales es recomendable que sea mayoritariamente integral, pero no consumir fibra insoluble como el salvado.
- Pastas (macarrones, espaguetis)
- Mariscos.
- Queso, requesón, leche, natas cremas, mantequilla.
-Prevención
- Consumo frecuente de agua o zumos y fibra soluble (de fruta) y dieta saludable.
- Se discute el papel de la vitamina A en el tratamiento del proceso. Como soporte vitamínico se suele utilizar el Aevit (acetato de retino) que favorece el desarrollo y el crecimiento del organismo. También se emplean vitaminas del complejo B: vitamina B6, vitamina B2. También otras vitaminas como la C, D, E y PP.
Tratamiento
El tratamiento de los pacientes con RP constituye uno de los graves problemas, todavía no resueltos, de la oftalmología. Se han utilizado diferentes tratamientos, normalmente basados en la posible causa de la enfermedad.
Ayudas ópticas
- Filtros para evitar deslumbramiento
- En el caso de agudeza visual y campos visuales reducidos, para leer se pueden utilizar las lupas y los circuitos cerrados de televisión. suelen leer mejor los caracteres blancos sobre fondo negro.
¿Qué es la Albinismo?Es una alteración genética que determina un fenotipo (apariencia física) muy característico, debido a la ausencia o hipo pigmentación en la piel, ojos y pelo. Es una condición metabólica poco habitual, debido a un defecto en el gen que se encarga de la síntesis y distribución de la melanina.
El albinismo causa un déficit de pigmentación en la retina, iris y coroides, lo que provoca que la pupila tenga un color rojo profundo y el iris color azul grisáceo o violáceo. La fóvea esta poco desarrollada por esta falta de pigmento, lo que limita la función visual y que la AV sea de 20/200
Epidemiología El albinismo tiene una incidencia mundial baja: una de cada 20.000 personas. Afecta del mismo modo a todas las razas. Las personas albinas se caracterizan por tener el pelo blanco o ligeramente dorado, la piel muy pálida o con un tono rosado, y los ojos azul violeta o rojizos, aunque la coloración rojiza la podemos observar al incidir la luz sobre los ojos, ya que al no existir pigmentación aparecen reflejados los vasos sanguíneos.
En la mayoría de los casos, el albinismo ocular viene del gen que se encuentra en la cromosoma X. Este tipo de albinismo ocular, ocurre exclusivamente en varones. La madre que tiene este gen se lo pasa a su hijo varón. Cada vez que una madre que lleva este gen da a luz, hay una probabilidad de 50% que su hijo tenga albinismo ocular. El albinismo ocular que viene de la cromosoma X también se conoce como albinismo ocular Nettleship-Falls.
Diagnostico Normalmente, se detecta por la apariencia de la persona, se confirma con un examen ocular completo realizado por un oftalmólogo y una evaluación genética junto con la investigación de la historia familiar.
Síntomas y signos -Agudeza visual reducida desde 20/60 a 20/400.
-Nistagmus - movimiento irregular y rápido de lado a lado de los ojos.
-Estrabismo
-Fotofobia. Sensibilidad a brillos de luces fuertes ó al resplandor.
Alteraciones oculares o síndromes asociados. Las personas con albinismo no son ciegos totalmente, tiene una visión bastante disminuida y como desenfocada, pero sí son capaces de distinguir formas y colores ya que sí poseen las células encargadas de estas funciones (conos y bastones) y en la mayoría de los casos pueden llevar una vida normal.
Medidas preventivas Al contrario de lo que se piensa las personas con albinismo también necesitan la luz y estar al aire libre como cualquier otra persona; por eso se les puede ayudar con gafas de sol con filtros especiales UVA y UVB y tener cuidado con las luces brillantes, que no enfoquen directamente a los ojos a la hora de trabajar o de la lectura.
Tratamiento A la hora de desarrollar actividades normales como la lectura o actividades cotidianas pueden necesitar el uso de gafas ya que algunos presentan astigmatismo o también hipermetropía. Hay muchas opciones para mejorar la visión como microscopios, telescopios, buscar libros con una versión de letra ampliada o macro tipos.
Ayudas ópticas -Microscopios
-Telescopios
-Filtros para evitar deslumbramiento
-CCTV
-Ayudas no ópticas: plumas negras, papel oscuro, guías para escribir, filtros y lámparas.
¿Qué es la Aniridia? Es una enfermedad del ojo congénita o hereditaria bilateral, el termino significa ausencia de iris, aunque existe un iris rudimentario, (falta de desarrollo o hipoplasia total o parcial), pudiendo acompañarse de hipoplasia de fóvea o nervio óptico causando un nistagmos congénito sensorial.
Epidemiología Es poco frecuente afecta a 1: 80.000-100.000 nacimientos, es de origen hereditario siguiendo un patrón de herencia autosómica dominante, o surgir de forma esporádica.
En España no se conocen estudios al respecto, pero se calcula un 1 caso por 80.000 o 100.000 personas.
Diagnostico El primer diagnóstico es visual ya que el recién nacido cierra los ojos en presencia de luz (fotofobia), es el principal signo de los afectados por esta enfermedad. El oftalmólogo debe revisar y determinar la gravedad así como las posibles alteraciones asociadas. Una vez diagnosticado, el paciente debe acudir a revisiones anuales.
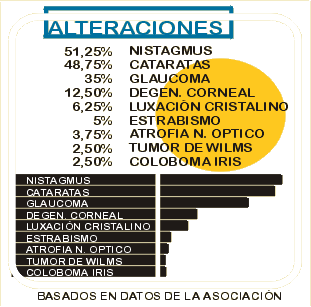
Síntomas y signos Baja visión: 20% o 10% en función de la lesión
Fotofobia
Queratinopatía (QAA): afecta al 20%. Se produce una sequedad ocular.
Glaucoma: afecta al 50-75 %
Cataratas congénitas: afecta al 50%.
Hipoplàsia macular y foveal.
Nistagmus.
Alteraciones oculares o síndromes asociados. Degeneración corneal
Hipoplasia del nervio óptico
Ambliopía
Síndrome de Gillespie
Estrabismo
Falta de pigmentación en la retina
Síndrome de Wilms
Retraso mental
Síndrome de WAGR
Medidas preventivas Evitar cambios bruscos de luz o contraluz
Más comodidad al sentarse de espaldas a una ventana para leer, pero con suficiente luz y sin reflejos sobre los cristales o espejos.
Utilizar gafas solares para evitar la fotofobia.
Tratamiento En la actualidad, no existe un tratamiento que englobe todos los síntomas de la aniridia, por lo que hay que tratarlo de forma individual. El primer paso, son las ayudas ópticas para mejorar la calidad de visión. También existen tratamientos farmacológicos para subsanar las queratinopatías (ojo seco), se pueden suministrar sueros para disminuir el dolor y el glaucoma, fármacos que disminuyen la presión intraocular.
Ayudas ópticas Filtros para evitar deslumbramiento
Tele – lupa
Atril.
Flexo con lupa y luz incorporada.
Libros adaptados con letra grande.
Programas específicos de ordenador.