La
catarata pediátrica es una de las principales causas
de ceguera infantil, afectando a 20.000 niños en todo el mundo. Pueden ser
congénitas cuando aparecen en el primer año de vida, desarrollarse durante la
infancia o ser traumáticas. Es de suma importancia su rápido diagnóstico y tratamiento para evitar
el desarrollo de ambliopía irreversible.
Los
últimos avances en técnicas quirúrgicas, material y diseño de las lentes
intraoculares, mayor comprensión de la neurobiología del desarrollo visual y el
uso temprano de lentes de contacto postoperatorias para la rehabilitación
visual han contribuido a mejorar los resultados de la cirugía de cataratas
pediátricas.
Además puede obtenerse un rápido diagnóstico con asesoramiento y
estudios genéticos en caso de cataratas hereditarias.
Sin
embargo, algunas cuestiones específicas de los ojos infantiles, como
inflamación postoperatoria, crecimiento axial después de la extracción, cálculo
de la potencia del implante, glaucoma secundario, opacificación de la cápsula
posterior y tratamiento de ambliopía siguen siendo un obstáculo para lograr
buenos resultados postoperatorios.
El examen del niño con cataratas
debe comenzar con una historia clínica detallada que incluya antecedentes
familiares, prenatales (abuso de drogas por parte de la madre o enfermedades
con fiebre y erupción) y del nacimiento, especialmente el peso ya que el bajo
peso podría estar asociado con cataratas congénitas bilaterales idiopáticas.
Deben descartarse etiologías metabólicas o sistémicas.
También
es importante saber cómo comenzaron las opacidades lenticulares, lateralidad y
progresión. Las cataratas unilaterales generalmente son aisladas, pero también
pueden estar asociadas con vasculatura fetal persistente y con otras
anormalidades oculares como lenticono/lentiglobo.
El
examen completo debería incluir biomicroscopía con lámpara de hendidura para
evaluar el tamaño, ubicación y densidad de la opacidad, alteraciones
capsulares, como defectos preexistentes posteriores y otras anormalidades
asociadas al segmento anterior.
Se
debe medir la presión intraocular y el diámetro de la córnea. Se realiza examen
de fondo de ojo en cataratas parciales y ultrasonido en cataratas totales, ya
que puede revelar anormalidades del segmento posterior que afectarían el
resultado visual. Debe controlarse la agudeza visual con los métodos adecuados
a las posibilidades del niño según su edad.
Cuando
los padres no están seguros acerca de cuándo comenzó la catarata, se puede
recurrir a fotos antiguas en las que el reflejo rojo puede dar la pauta de la
presencia de cataratas durante determinado período crítico en el desarrollo
visual. También puede examinarse con lámpara de hendidura a padres y hermanos,
para investigar alteraciones no diagnosticadas que indicarían una causa
hereditaria para la catarata infantil.
Según
la morfología las cataratas pediátricas pueden clasificarse como cataratas que
abarcan todo el cristalino, cataratas centrales, anteriores, posteriores,
opacidades punteadas del cristalino, cataratas coralinas, asociadas a
vasculatura fetal persistente, etc. En el presente estudio, queremos destacar
las cataratas con una morfología típica específica de ciertas patologías
sistémicas o síndromes, lo que puede ayudar en el diagnóstico.
Las
cataratas totales pueden ser esporádicas o hereditarias, se pueden observar en
casos de síndrome de Down y síndrome de rubeola congénita. Una rápida
intervención quirúrgica es fundamental para evitar el desarrollo de ambliopía.
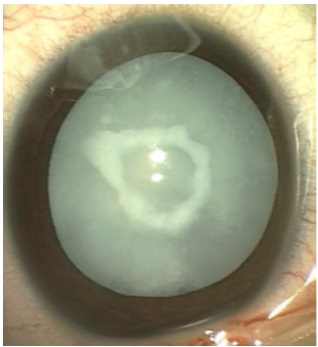
Fig. 1 Catarata total con placa capsular
anterior central.
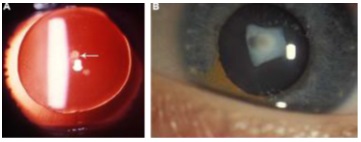
La catarata polar anterior no es característica de
ninguna patología en particular, pero normalmente se observa en pacientes con
aniridia (Fig 2 A). Pueden tener forma de puntos, placas o en pirámide. La
piramidal es la forma más grave de catarata anterior el vértice de la pirámide
ingresa en la cámara anterior y también se ha observado en niños con
retinoblastoma y síndrome de Ehlers Danlos (Fig. 2B).
La catarata anterior subcapsular está asociada con
uveítis, trauma, irradiación y dermatitis atópica. El lenticono anterior es una
patología bilateral que se presenta en casos de síndrome de Alport y rara vez
con síndrome de Waardenburg.
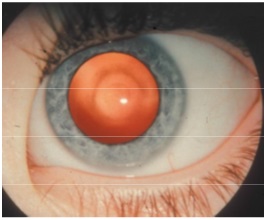
La catarata en gota de aceite es una opacidad nuclear
que suele verse en niños con galactosemia (Fig. 3), quienes también pueden
presentar catarata posterior subcapsular o una opacificación pequeña nuclear y
cortical. Estos cambios son reversibles con una modificación rápida de la dieta.
La catarata en girasol es un tipo de catarata subcapsular anterior que
suele verse en casos de enfermedad de Wilson, patología que afecta el
metabolismo de cobre y que produce su acumulación en el hígado y ganglios
basales. También es reversible con tratamiento con penicilamina.
La catarata subcapsular posterior puede ser inducida por
drogas (generalmente esteroides), o deberse a una complicación por tratamiento
de radiación de tumores oculares o perioculares. También se ha observado en
casos de síndrome de Turner, enfermedad de Fabry, síndrome Bardet.Biedel y
neurofibromatosis tipo 2.
La catarata membranosa, opacidad en forma de
disco es típica del síndrome de Hallermann-Streiff y también se ha informado en
niños con rubeola congénita, síndrome de Lowe y vasculatura fetal persistente.
La catarata en forma de cuña es característica del
síndrome de Stickler , así como síndrome Conradi Hünermann,
neurofibromatosis tipo 2 y enfermedad de Fabry.
La catarata sutural compromete la sutura Y del cristalino. Se suelen
descubrir casualmente en un examen de rutina. Se han descripto casos en mujeres
portadoras del síndrome Nance-oran y en los hombres se presenta con cataratas
densas, visualmente significativas.
Fig. 2 Catarata polar anterior
Fig. 3 Catarata en gota de aceite, caso de
galactosemia
Conclusiones: El pronto diagnóstico e
intervención en casos de catarata pediátrica es esencial para prevenir
ambliopía irreversible. Es fundamental un examen ocular completo, incluyendo comienzo,
duración y morfología de la catarata. Una evaluación de la salud general del
niño, por parte de un pediatra, sirve para descartar patologías sistémicas
asociadas.
Síntesis
y traducción: Dr. Martin Mocorrea, editor responsable de Intramed en la especialidad
de oftalmología
1. Foster A, Gilbert C, Rahi J.
Epidemiology of cataract in childhood: a global perspective. J Cataract Refract
Surg. 1997;23 Suppl 1:601–604.
2. Holmes JM, Leske DA, Burke JP, Hodge DO. Birth
prevalence of visu¬ally significant infantile cataract in a defined US
population. Ophthalmic Epidemiol. 2003;10(2):67–74.
3. Stayte M, Reeves B, Wortham C. Ocular and
vision defects in preschool children. Br J Ophthalmol. 1993;77(4):228–232.
4. Lee KA, Park MH, Kim YJ, Chun SH. Isolated
congenital hereditary cataract in a dizygotic twin: prenatal ultrasonographic
diagnosis. Twin Res Hum Genet. 2013;16(5):994–997.
5. Vasavada AR, Raj SM, Nihalani B. Rate of axial
growth after congenital cataract surgery. Am J Ophthalmol. 2004;138(6):915–924.
6. Jasman AA, Shaharuddin B, Noor RA, Ismail S,
Ghani ZA, Embong Z. Prediction error and accuracy of intraocular lens power
calculation in pediatric patient comparing SRK II and Pediatric IOL Calculator.
BMC Ophthalmol. 2010;10:20